VII. Electrophoresis
A. Macromolecule is accelerated by a force (like sedimentation)
- F = q E
[q is the net charge on the molecule;
E is the electric field
strength experienced by the molecule]
- This causes an acceleration until the velocity,
v, causes a frictional force
equal to but opposite in direction to the applied force F = q E = f v
- Like sedimentation, we can define a mobility per unit field,
U
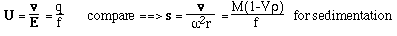
- For a spherical molecule of radius, r, and charge z e (e = elementatry charge,
the charge on 1 electron)

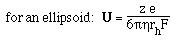
F is the Perrin shape factor; rh
is the radius of a sphere of equal volume.
B. Rigorous quantitative treatment is very difficult because the electric
field actually felt by the macromolecule is difficult to evaluate due to the
fact that the macromolecule is a very large ion in solution with many small
counterions. 2 extreme situations:
- Very Low Ionic Strength
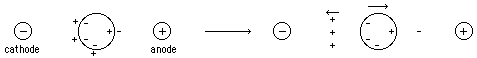
Once the macromolecule is separated slightly from its
counterions, it takes enormous energy to pull them further apart ==> charge
separation counteracts the external field resulting in little or no molecular
transport.
- Very High Ionic Strength --
overcomes the problem of charge separation (the macromolecule will always have
enough counterions around). But this creates an ion cloud around the particle
partially shielding it from the external field. This does not prevent
electrophoretic movement, but it does complicate rigorous analytical
treatment.
- Most electrophoretic experiments (whether preparative or
analytical) are analyzed semi-empirically.
C. Experimental Approaches -- Boundary and Zonal (just like
sedimentaiton)
- Boundary: Analogous to Boundary
Sedimentation -- measure the rate of movement of the boundary and calculate
U from E and v -- hardly ever used anymore
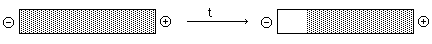
- Zonal: Analogous to Zonal
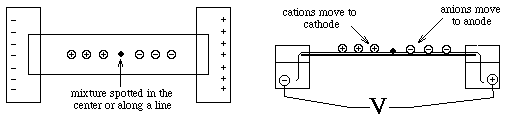
Sedimentation in density gradients
- sample is applied in a zone (small region: a spot on moistened
paper or a band on a gel) and an applied field causes the molecules to
separate into zones based upon different U's.
- Like Zonal sedimentation, need some way of stabilizing the zones to
prevent mechanical mixing (from vibrations) or convection mixing (from
temperature differences -- a particularly severe problem with resistance
heating caused by the electric field).
- Paper Electrophoresis:
- a strip of paper is kept moist with buffer to make it
electrically conductive; ends are dipped into buffer solutions containing
electrodes across which an electric potential is applied
- Used primarily for separation of small molecules ==> must use a high
voltage, otherwise they diffuse too rapidly ==> paper must be cooled
(usually by water)
D. Gel Electrophoresis Media -- Three types
- Starch Gel -- swollen potato
starch granules (little used now except for prep isoelectric focusing)
- Agarose Gel -- purified large MW
polysaccharide (from agar) ==> very open (large pore) gel used frequently
for large DNA molecules
- Polyacrylamide Gels -- most
commonly used gel because they are very stable and can be made at a wide
variety of concentrations or even with a gradient of concentrations ==>
large variety of pore sizes

- Acrylamide Concentrations --
typically 5-20% by weight (5%, 7.5%, 10%, 12.5%, 15%, 20% are commonly used
values) ==> gel is mostly water. Acrylamide polymerizes in head-to-tail
fashion to form long polymers which form a complex network held together by
bis-acrylamide crosslinks. The cris-crossing polymers create pores in the gel;
the size of pores is determined by the acrylamide concentraion.
- Acrylamide can be polymerized into any desired shape -- two
shapes used for electrophoresis
- Tube Gels -- polymerize in glass tubing ==> cylindrical
shape
- Slab Gels -- polymerize between glass plates
G. Nucleic Acids -- How does a gel affect mobility?
Let's consider simple (conformationally) molecules moving free
in solution in the presence of an electric field.
- Structure -- Random coil
of...
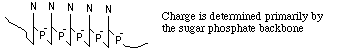
Thus: Z is proportional to
M, the molecular weight
Recall that for an
ellipsoid: f = 6 p h rh F and: 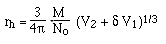
==> f is proportional to
M (for molecules with similar shapes)
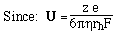 --> both numerator and denomenator contain terms which are
directly proportional to M ==> M terms cancel out and the electrophoretic mobility, U, should be independent of the
size of the nucleic acid.
--> both numerator and denomenator contain terms which are
directly proportional to M ==> M terms cancel out and the electrophoretic mobility, U, should be independent of the
size of the nucleic acid.
N. Davidson at CalTech
measured U for nucleic acids
without a supporting gel, and found that it is constant from 1 nucleotide up
to 1.7 x 105
nucleotides.
- How can we use electrophoresis to separate molecules?
Electrophoresis in a gel matrix -- the gel sieves the molecules
- large molecules move slowly because they have difficulty going
through the pore
- small molecules move rapidly, their freedon is not restricted
- from Physical Chemistry:
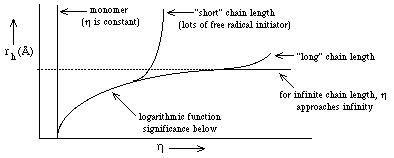
- a. h becomes a function of the particle
size (rh) because large particles "feel" the gel matrix more than
small particles--they more often come in contact with the gel matrix. Thus,
since f = 6 p h rh ==> h is larger when rh is large and f increases ==> U decreases
because
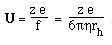
- for a 7.5% polyacrylamide gel--average pore size is ~50Å
- How is pore size related to gel concentration?
- s1 =
pore size for gel concentration 1 -- c1
- s2 =
pore size for gel concentration 2 -- c2
- d = diameter of the hydrated polymer
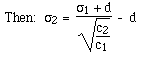
e.g. for a 7.5% gel s1 = 50Å =; d = 10 Å ==>
for a 30% gel: s2 = 20Å
Thus, we can adjust the gel concentration
to produce a pore size appropriate for the sizes of molecules we wish to
separate.
- U is measured as the "relative"
distance traveled, Rf = R / Rmax
Find
that Rf depends upon log
M ==> Rf = b - a log M where "a" and "b" are
constants determined (primarily) by the gel; they're measured empirically by
plotting Rf for molecules of known M and making a
calibration curve. Note: the relationship is linear only over a limited range
of log M determined by the pore size of the gel and by the polymer length.
This is because h is a
logarithmic function of molecule radius, rh over a limited range (see above).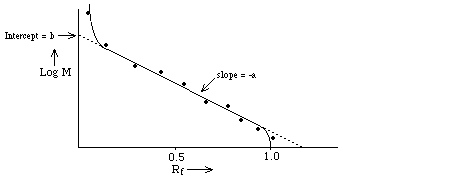
- Choice of Gel
- Agarose: 0.2 % for Nucleic Acids up to M = 150 x
106
0.8 % for Nucleic Acids up to M = 50 x 106
- Polyacrylamide: for smaller Nucleic Acids; choose % acrylamide to
produce correct size pores
H. Electrophoresis of Proteins
- Less straightforward because charge on proteins is much more
variable -- it depends upon:
- amino acids composition
- pH;
- net charge can be + or - or 0
- Electrophoresis of "native" proteins is relatively rare
except for Isoelectric Focusing described later. What is needed is a way of
modifying proteins so that z is proportional to M as is the case with nucleic
acids.
- SDS PolyAcrylamide Gel Electrophoresis -- SDS
PAGE
- Sodium Dodecyl Sulfate = Sodium Lauryl Sulfate:
CH3(CH2)11SO3-
Na+
This is a detergent because it contains a
hydrophobic region, the CH3(CH2)11 tail,
attached to a hydrophilic group, SO3- Na+,
making it amphipathic. It is a very strong detergent which denatures
proteins by binding to the polypeptide backbone.
- Measurements show that most proteins bind 1.4 gm SDS/gm protein
with very little variation (except for some membrane proteins; membrane
proteins are very hydrophobic and may bind more SDS) ==> The charge
on an SDS-protein complex is determined almost entirely by SDS --> -1
charge for each SDS. Since the amount of SDS bound is determined by the size
of the protein and all protein/SDS micelles are anionic ==> z is
directly proportional to M
- Hydrodynamic studies show that the shape of an SDS/protein complex is a
rod or prolate ellipsoid--called a micelle--of ~18Å diameter and a length
proportional to M
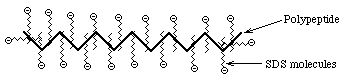
- U in solution is independent of M, just like Nucleic Acids
- Thus, electrophoresis of SDS/protein micelles through a polyacrylamide
gel should separate them according to M as with nucleic acids ==>
Rf = b - a log M
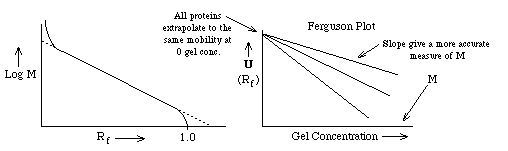
- If one plots the mobility for different proteins as a function of gel
concentration, a Ferguson Plot, one finds that the mobilities of all
proteins extrapolate to the same value at [gel conc.] = 0 as expected. The
slope of the mobility of a single protein provides another more accurate
method of estimating its molecular weight from SDS PAGE.
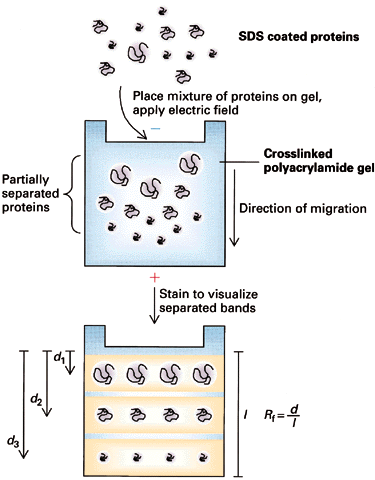
I. Discontinuous Electrophoresis (Disc): Most common type of SDS-PAGE
- Buffers--2 layers:
upper layer contains low
mobility ions; lower
layer contains high mobility
ions
- The two zones move down with a sharp boundry between them.
Why? Faster lower buffer ions should move faster leaving slow ones
behind.
- They experience different potentials: VU and VL
- Ohms Law: V = R i: i = current and is the same for both layers, R
is the resistance. The resistance of the upper zone, RU, is
greater than the resistance of the lower zone, RL,
because:

where
ci = concentration;
mi = mobility;
zi= charge on the ith
ion. ==> RU will be
larger than RL because
mU is smaller than
mL
- Thus, if cU and
cL are chosen
appropriately, VU will
be enough larger than VL to compensate for lower mobility of ions in the upper
zone ==> 2 zones move at the same speed with a sharp boundry between
them.
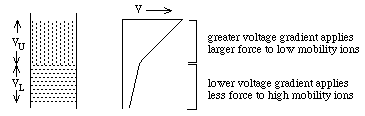 .
.
- Why a sharp boundry? (1) if a mobile lower zone ion drifts into
the upper zone, it immediately experiences a higher potential,
VU, and speeds up until it reaches the lower zone where the lower
potential, VL, causes it to slow down again. (2) if a low
mobility upper zone ion drifts into the lower zone, it immediately
experiences a lower potential, VL, which causes it to slow down
until it drifts back into the upper zone.
- What about proteins? Choose
upper and lower zone ions so that mU < mproteins < mL
==> proteins will be concentrated
at the interface into thin zones stacked in order of protein mobility. Note:
all this assumes electrophoresis in a "stacking" gel with large pores which do not inhibit
protein movement.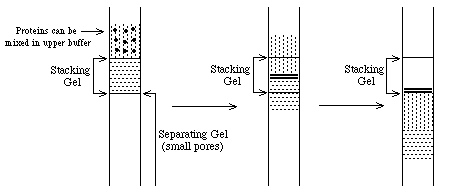
When the thin bands of proteins reach the separating gel, their
mobility decreases dramatically and the upper buffer ions pass them. At this
point, the proteins are separated according to their individual mobilities
(sizes). The whole point of this buffer system is to concentrate the proteins
into very thin zones before separating them.
J. IsoElectric Focusing:
All protein charges vary from a net positive charge at low pH
(-COOH and -NH3+ forms of acidic and basic functional
groups) through 0 at some intermediate pH to a net negative charge
(-COO- and
-NH2 forms) at high
pH.
- pI - IsoElectric Point: pH at
which a protein has a net 0 charge (positive and negative charges balance).
Depends mostly on the amino acid composition and a little on the tertiary
structure
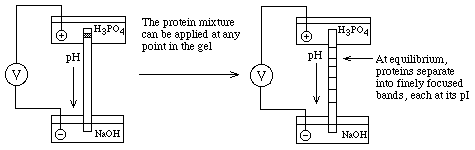
- Create a pH gradient in a gel:
Can be done on a slab (vertical or horizontal) or a tube.
This is analogous
to Equilibrium Density Gradient Centrifugation (IsoPycnic Centrifugation); the
final positions of each band depend only on an intrinsic property of the
proteins, their pI's, and not on where they started in the gel
- How to make a stable pH gradient? Must have a buffer for
each pH along the gradient
==> Ampholytes (various commercial names) are
small organic molecules with different combinations of acidic and basic groups
so that each one has a different pKa. If one electrophoreses a mixture of
ampholytes (polyampholytes) with H3PO4 in the Anode buffer reservoir (to buffer
it at very low pH) and NaOH in the Cathode buffer reservoir (to buffer it at
very high pH), each ampholyte will migrate to a pH equal to its pKa and buffer
the pH at that point.
K. Two-Dimensional Electrophoresis: There are many variations; all basically
combine 2 types of electrophoresis
- Most common combines Isoelectric Focusing in tube gels and
SDS-PAGE in slab gels

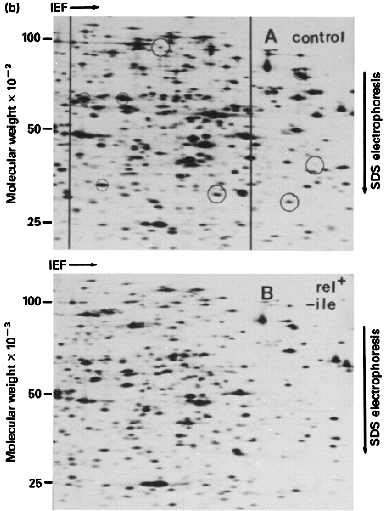
Final gel
has a complex mixture or proteins separated by pI along the horizontal axis
and by log M along the vertical axis. Can resolve thousands of spots
(proteins) by this technique. Analysis is now automated by computer so that
one can do 2D gels on whole cell extracts and monitor how each protein changes
during: a) Development; b) Transformation; c) Excitation -- e.g. by a hormone
etc.
- Other Variations
- SDS-PAGE -- non-reducing conditions (-S--S- bonds intact) +
reducing conditions
- SDS-PAGE -- proteins X-linked + X-links broken
- Native Gel (non-denatured proteins ) + SDS-PAGE
VIII. pH Measurement
A. Glass Electrode

- H+
inside have a higher chemical potential, µH+, than H+ outside because their concentration is
higher. The difference in µH+ (free energy due to different concentrations) is given by the
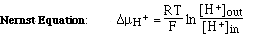
F is called
the Faraday const. and
converts to Volts
- H+
diffuse out of the bulb creating a charge separation or voltage across the
glass, DV.
H+ ions
will continue to diffuse across the glass until:
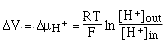
==> if
we can measure ÆV we can detemine [H+]out since we already know R, T, F and
[H+]in

- Calomel Electrode -- a
convenient reference electrode; ÆV across the glass electrode is compared with
the reference ==> reference electrode must be pH independent. KCl
is used to make contact between the solution and the reference electrode
through a fiber capillary or a fibrous plug.
- Other Types of Reference Electrodes
- Ag-AgCl -- for high temperatures
- Hg-HgSO4 -- for Cl-free electrode
- Combination Electrode -- Glass
electrode + reference electrode combined into one unit --> more compact for
measuring pH of small volumes.6. In addition to the ÆV's across the glass
electrode and across the reference electrode, there are also ÆV's
across:
- the AgCl-coated wire in the glass electrode;
- an asymmetry potential across the H+-permeable glass when the pH is the
same on both sides
- liquid-junction potential of the reference electrode due to the fact
that KCl is used to conduct current and K+ and Cl- don't diffuse at the same
rate. Fortunately, all of these are pH-independent (like the reference
electrode) and can be corrected by calibration.
B. Complications of pH measurement
- Dependence on concentrations of ions
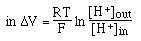
one is
really measuring the H+
activity. This is related to concentrations and is approximately equal to
concentration in dilute solutions but can be significantly different than
concentration when concentrations of other ions are very high ==> measure
pH at approximately the same concentrations as those at which the buffer will
be used or take the change due to dilution into account.
- Electrode contamination
- foreign material -- e.g. protein film -- may coat the glass
electrode -->should remove with detergent or acid
- Tris reacts with some commercial electrodes --> see that your
electrode gives correct results with Tris if you use this buffer.
- Sodium Error: many glass
electrodes are somewhat permeable to Na+ ==> pH can appear to be lower
([H+] greater) than it
actually is! Most noticeable at high pH where the error can be as much as 1-2
pH units
- use special Na+ impermeable
electrodes when measuring high pH's of buffers containing high
concentrations of Na+
- use K+ salts instead of Na+ salts
- Temperature: there are 2
variations with temperature
- in Nernst Equation:
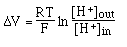 <-- this
temperature variation is corrected with the
<-- this
temperature variation is corrected with the
temperature compensation
control on the pH meter.
- buffer equilibrium may shift with a change in temperature -- this will
be different for different buffers and can go in different directions
depending upon whether dissociation is exothermic or endothermic. This is a
problem with Tris buffers whose pH decrease approximately 0.03 pH units for
every degree change from 25°C --> 5°C.
C. Ion Specific Electrodes:
can be constructed for other ions by changing the composition
of the glass in the glass electrode. The principles of operation will be exactly
the same as the pH electrode. There are 3 basic ways of altering the
electrode.
- Change the glass:
- SiO2, Na2O, CaO ==> H+ electrode
- SiO2, Al2O3,Li2O ==> Na+
electrode
- SiO2 + other components ==> Li+, K+, Rb+, Cs+, Ag+, Cu+,
Ti+, NH4+ electrodes-
- Thinly Sliced Crystals:
- LaF3 ==> F- electrode
- Ag2S ==> S-2 electrode
- AgX ==> X- electrode (X is a halide)
- Liquid Membrane (inorganic salt
dissolved in an organic solvent and held in porous glass or plastic:
Ca(C10H21O)PO2 ==> Ca++electrode